This chapter originally appeared in Characterization and Failure Analysis of Plastics, published by ASM in 2003
and a revision in ASM Handbook, Volume 11B, in 2023
The body of the chapters are similar, but the revision contains completely new case studies.
The ultimate objective of a failure analysis is to ascertain the mode and the cause of the failure, regardless of the material from which the part was fabricated. The investigation is performed in generally the same manner, whether the failed component was produced from metal or plastic or a combination of these materials. Thus, the general steps required to conduct a comprehensive failure investigation are the same. In general, the failure analysis process is analogous to putting together a jigsaw puzzle. A failure analysis requires assembling bits of information into a coherent and accurate portrayal of how and why the part failed. Reaching the objectives of the plastic failure analysis, namely, the determination of the mode and cause of the failure, or expressed alternatively, evaluating how the part failed, and why it failed, requires a scientific approach and a broad knowledge of polymeric materials.
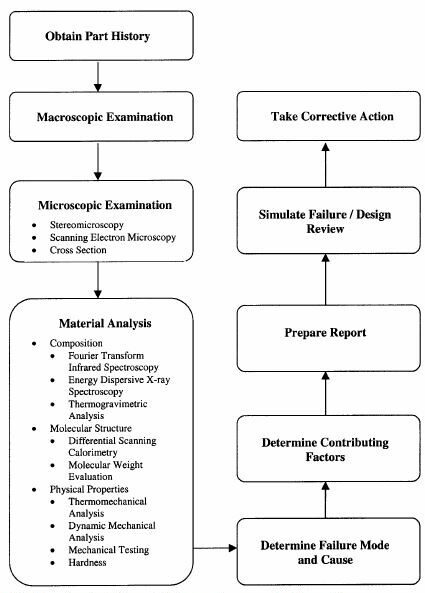
Steps for performing failure analysis. The steps are the same regardless of the material.
Failure within plastic components can occur in several different ways, including esthetic alteration, deformation/distortion, degradation, wear, and fracture. In the case of failure involving fracture, there are a number of common plastic failure mechanisms, such as, such as brittle fracture, ductile overload, creep rupture, environmental stress cracking, molecular degradation, and fatigue. In the case of failure involving fracture, the determination of the failure mode involves identifying how the crack initiated and how it subsequently extended. This is usually ascertained using a number of visual based techniques, such as stereomicroscopy, scanning electron microscopy (SEM), and the preparation of mounted cross sections. Noncatastrophic failure modes are also relevant, and these include discoloration, distortion, and contamination. Assessing the mode of the failure is often not as difficult as establishing why the part failed. Evaluating why the part failed usually requires analytical testing beyond the visual-based techniques. In many cases, a single cause cannot be identified, because multiple integrated factors may have contributed to the failure. All the factors that affect the performance of a plastic component can be classified into one of four categories: material, design, processing, and service conditions. These factors do not act independently on the component, but instead act in concert to determine the performance properties of a plastic component.
The principal differences between how failure analyses are performed on metal and plastic materials center on the techniques used to evaluate the composition and structure of the material. Unlike metals, polymers have a molecular structure that includes characteristics such as molecular weight, crystallinity, and orientation, and this has a significant impact on the properties of the molded article. Additionally, plastic resins usually contain additives, such as reinforcing fillers, plasticizers, colorants, antidegradants, and process aids. It is this combination of molecular structure and complex formulation that requires specialized testing. While the chemical composition of a failed metal component can often be evaluated using a single spectroscopic technique, a similar determination requires multiple analytical approaches for a part produced from a plastic resin.
This article reviews those analytical techniques most commonly used in plastic component failure analysis. The highlighted techniques include:
- Fourier transform infrared spectroscopy (FTIR)
- Differential scanning calorimetry (DSC)
- Thermogravimetric analysis (TGA)
- Thermomechanical analysis (TMA)
- Dynamic mechanical analysis (DMA)
- Gel permeation chromatography (GPC)
- Melt flow rate (MFR)
- Intrinsic viscosity (IV)
- Mechanical testing
- Energy dispersive X-ray spectroscopy (EDS)
- Scanning electron microscopy (SEM)
The description of the techniques is not designed to be a comprehensive review and tutorial, but instead is intended to make the reader familiar with the general principles and benefits of the methodologies. The descriptions of the analytical techniques are supplemented by a series of case studies. The technique descriptions refer to the case studies, and the two are written in a complimentary manner to illustrate the significance of the method. The case studies also include pertinent visual examination results and the corresponding images that aided in the characterization of the failures.
The body of the chapters are similar, but the revision contains completely new case studies.